Predicting protein-protein interactions
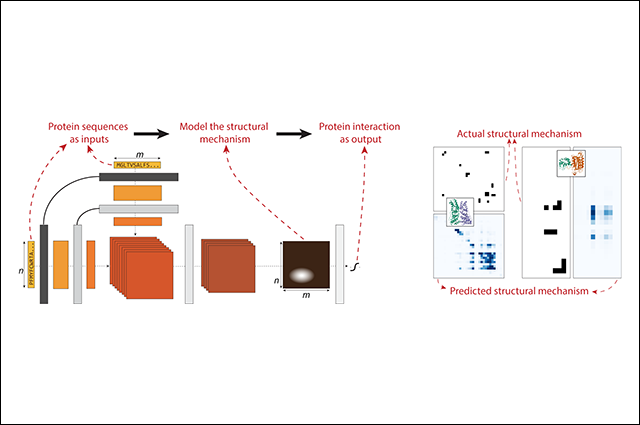
In research published in the journal Cell Systems, Professor Lenore Cowen of the Tufts Department of Computer Science and colleagues from Massachusetts Institute of Technology (MIT) collaborated to design a structurally-motivated deep learning method built from recent advances in neural language modeling. The team’s deep-learning model, called D-SCRIPT, was able to predict protein-protein interactions (PPIs) from primary amino acid sequences.
Those predictions allow researchers to model PPI networks with a clustering method and enable the detection of functional subnetworks, or modules. Scientists study organisms’ PPI networks as a means of understanding their signaling circuitry, which could lead to better prediction of cell behavior and gene functions, while finding functional modules in PPI networks could help researchers reach stronger understandings of cellular functional organization.
Cowen along with researchers Sam Sledzieski, Rohit Singh, and renowned computational biologist Bonnie Berger from MIT’s Computer Science and Artificial Intelligence Lab found that the D-SCRIPT model, trained on more than 38,000 human PPIs, was better able to generalize when compared to the current state-of-the-art approach (the deep-learning method PIPR), and therefore could characterize fly proteins. They also applied D-SCRIPT to screen for PPIs related to cow digestion and identified functional gene modules that related to immune response and metabolism.
The researchers concluded that the D-SCRIPT model trained on human PPI data could be applied to many species of interest – critically, even those that have been rarely studied or that lack PPI data.
Read more in Cell Systems: D-SCRIPT translates genome to phenome with sequence-based, structure-aware, genome-scale predictions of protein-protein interactions
Department:
Computer Science